:
Home
About ReSite
Download
How To Use
Contact Us
Department of Pharmacoinformatics
NIPER, SAS Nagar
NIPER, SAS Nagar


Molecular Docking is a computer algorithm which predicts the preferred orientation of a rigid or
flexible ligand into the binding site of the receptor and predicts the binding affinity of the complex
based on scoring functions. For understanding the structural principles that determines the strength
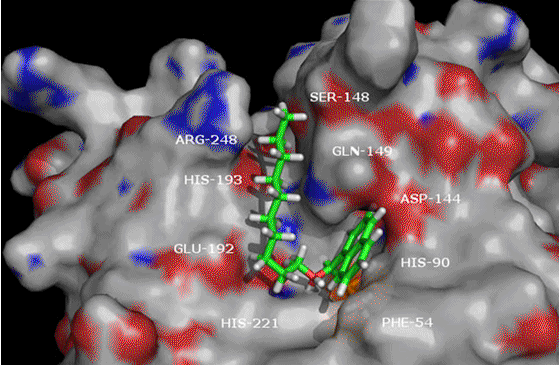
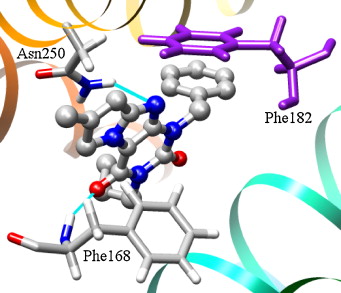
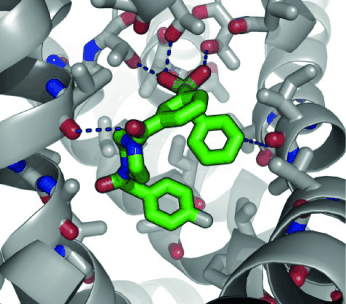
of a protein-ligand complex, the ability to acknowledge the residues involved in binding are required.
As it is often tedious to visualize the surrounding active site residues of the various docking poses of
each and every ligand, there is a need for a quick , textual presentation of these residues.
Here we present ReSite-a tool developed at Department of Pharmacoinformatics, NIPER
(S.A.S. Nagar) that focuses on reporting all the residues present within a distance constraint in the
active site playing a key role in protein-ligand interactions.


A Tool for analysis of the active site residues in protein-ligand interactions
DISCLAIMER: The use of this tool is intended for research purposes only and not for any clinical or commercial use. It is a non-profit service to the academic and non-academic scientific community. It is limited to applying best efforts in providing a useful service. These pages may provide links to information provided by external services which are not in any way under our control. We cannot, therefore, be held responsible for the content or accuracy of external pages.